Anthropic опубликовал свежее исследование, и оно ломает привычное представление о том, на что способны современные LLM в науке. Новый бенчмарк BioMysteryBench проверяет, может ли модель решать реальные задачи биоинформатики, и Claude уже на равных с PhD-экспертами, а на части задач обходит целые панели учёных.
Зачем вообще понадобился ещё один бенчмарк? Существующие тесты вроде MMLU-Pro, GPQA и LAB-Bench меряют знания и рассуждения, а BLADE, BixBench и SciGym пробуют оценить агентные сценарии. Но настоящая наука это шумные данные, субъективные методические решения и куча задач, которые человечество пока не решило. Anthropic собрал 99 заданий от доменных экспертов, причём правильный ответ выводится не из субъективного вывода учёного, а из контролируемых свойств данных или валидированной метаинформации (например, ответ подтверждён ПЦР-анализом).
Claude получает доступ к каноническим биоинформатическим инструментам, может ставить пакеты через pip и conda, дёргать NCBI и Ensembl, скачивать референсные геномы. Решения оцениваются по итоговому ответу, а не по пути к нему. Это даёт модели свободу выбирать стратегию: WGS-анализ, scRNA-seq, ChIP-seq, метилирование, метагеномика, протеомика, метаболомика.
Что в итоге: на 76 задачах, которые смог решить хотя бы один человек, последние поколения Claude уверенно выходят на уровень доменных экспертов. На 23 задачах, которые панель из пяти PhD не осилила, Claude Sonnet 4.6 и более старшие модели решают значимую долю, а Claude Mythos Preview добирается до 30 процентов. Для контекста: это вопросы, на которых группы профильных учёных просто пасуют.
Самое интересное это стратегии. Иногда Claude идёт по человеческой тропе, иногда вообще по другой. Например, там где эксперт запускал алгоритм или базу для аннотации, модель просто узнаёт паттерн в последовательности по памяти. Авторы напоминают, что первый эукариотический промотор открыли так же, заметив повторяющееся TATA. У LLM этот тип интуиции потенциально работает в гигантских масштабах.
Anthropic выделяет два ключевых приёма Claude. Первый, know-it-all: модель тащит из своей базы знания о структурной биологии, молекулярных профилях и метаанализе сотен тысяч статей и комбинирует это с живым анализом данных. Второй приём полезен и людям: когда модель не уверена, она запускает несколько разных методов и берёт ответ, на котором сходятся независимые подходы.
Есть нюанс с надёжностью. Claude Mythos Preview сам провёл анализ своих результатов и заметил, что на человеко-решаемых задачах модель бимодальна: либо решает 4 или 5 раз из 5, либо никогда. На сложных задачах распределение размазывается, почти половина побед это удачные попадания, а не воспроизводимый метод. То есть разрыв в точности между лёгкими и сложными задачами это только верхушка, под ним лежит более интересная проблема стабильности рассуждений.
Параллельно Genentech и Roche выкатили CompBioBench на 100 задач вычислительной биологии. Картина та же: Claude Opus 4.6 берёт 81 процент в общем зачёте и 69 процентов на самых сложных вопросах. Фронтирные модели реально становятся рабочими коллабораторами для биоинформатики, а не просто болталками.
Если коротко, граница того, что AI может в науке, сместилась. Модели уже не догоняют учёных в биоинформатике, на отдельных задачах они впереди. И главный вопрос теперь не «пройдёт ли модель экзамен», а «решит ли она проблему, которую люди не решили за десятилетия».
Реальное время, прозрачные рассуждения и открытые веса — ускоряя исследования по всей планете
🔭 Вклад в мировую науку: Grok — серия моделей от xAI (Илона Маска), ориентированная на максимальную откровенность и доступ к реальному времени. В отличие от других LLM, Grok использует живые данные из X (Twitter) для актуальных научных обзоров. Базовая модель Grok-1 (314B параметров) выпущена с открытым весом, что дало толчок тысячам исследователей в области масштабируемых архитектур, а Grok-1.5V привнёс мультимодальность в обработку научных изображений.
🏆 Научные достижения Grok
⚡
Grok-1: Открытая архитектура 314B
В марте 2024 года xAI выпустила веса Grok-1 — гигантской модели с 314 миллиардами параметров, одной из крупнейших open-source моделей. Это позволило учёным изучать внутренние механизмы масштабных MoE-подобных архитектур и проводить эксперименты без ограничений API.
🔥 Более 50 000 загрузок за первую неделю
🌐
Доступ к научным данным в реальном времени
Благодаря интеграции с X (Twitter), Grok может анализировать свежие научные препринты, обсуждения конференций и данные сенсоров в реальном времени. Это ускорило мониторинг эпидемий, космической погоды и быстропрогрессирующих областей (например, Room Temperature Superconductors).
📡 real-time научный анализ
🧠
Прозрачные цепочки рассуждений
Grok разработан с фокусом на объяснимость: модель показывает логические шаги, что критически важно для проверяемости научных гипотез. Многие лаборатории используют Grok для обучения студентов основам вывода и верификации доказательств.
📖 Повышение воспроизводимости
🖼️
Grok-1.5V: Мультимодальность для науки
Модель способна интерпретировать сложные схемы, графики, рентгеновские снимки и диаграммы из научных статей. Учёты в области материаловедения и биологии используют Grok-1.5V для автоматического извлечения данных из изображений.
🏥 Точность 91% на научных визуальных бенчмарках
314B
параметров (крупнейшая открытая)
500+
научных публикаций с Grok
150+
университетов используют Grok API
24/7
режим real-time научных данных
* Данные на основе репозиториев, arXiv и отчётов xAI (2024-2025)
“Grok-1 стал первым по-настоящему открытым гигантом в мире LLM. Возможность исследовать внутренности 314B модели дала сообществу бесценные данные о поведении масштабных сетей. А интеграция с живыми потоками X (Twitter) открыла новую эру — «науку в реальном времени».”
— Проф. Эндрю Нг, AI Fund / Stanford (адаптированная оценка открытых моделей)
⚙️ Уникальные научные инновации
🔭
Астрофизика и космос
Grok активно используется в обработке данных телескопов (включая проекты Илона Маска). Благодаря способности анализировать многомерные ряды и изображения, модель помогает идентифицировать аномалии в данных обсерваторий и прогнозировать солнечные вспышки.
🧬
Анализ научных текстов
Способность Grok работать с огромными контекстами (128K токенов) позволяет учёным загружать целые диссертации и получать мгновенные рецензии, выделять противоречия и генерировать мета-обзоры.
📑 Автоматическое рецензирование
🧪
Реактивность научных гипотез
Grok может выдвигать контр-примеры и находить логические ошибки в рассуждениях, что помогает исследователям улучшать формулировки теорий и избегать когнитивных искажений.
📊
Интеграция с инструментами Python
Модель генерирует код для научных вычислений (NumPy, SciPy, Matplotlib) и может самостоятельно исполнять его через интерпретатор. Это ускоряет обработку экспериментальных данных в физике, химии и биоинформатике.
📅 Хронология влияния Grok на науку
Ноябрь 2023 — Анонс Grok-1 Первый LLM с реальным доступом к X, вызвавший волну исследований в области “real-time prompting” и семантического анализа социальных потоков для научных прогнозов.
Март 2024 — Открытие весов Grok-1 (314B) Революционное событие: самая большая open-source модель на тот момент. Тысячи исследователей начали анализировать её внутренние представления, что породило >200 статей на arXiv за 3 месяца.
Апрель 2024 — Grok-1.5V (мультимодальный) Первая vision-модель от xAI. Научные команды интегрируют её в анализ медицинских изображений и спутниковых снимков.
2025 — Grok в научных кластерах Расширенный контекст и инструментарий: использование Grok для симуляций в астрофизике, климатологии и геномике. Сообщество отмечает рост воспроизводимости экспериментов благодаря открытости.
📡 Открытость + скорость
xAI поставила во главу угла максимальную прозрачность и доступ к реальным данным. Grok-1 (314B) с открытым весом стал платформой для исследований масштабируемых AI-систем. Учёты из 70+ стран используют Grok для обработки научных потоков в реальном времени, а открытые отчёты xAI помогают развивать Explainable AI.
🔓 Веса Grok-1 доступны на GitHub
🎓 Научные партнёры: Harvard, Caltech, MIT, University of Toronto, Московский государственный университет
📈
Grok в научных публикациях и проектах
С момента выпуска Grok-1 количество ссылок на модель в рецензируемых журналах выросло на 400% (Q1 2025). Особенно сильное влияние в области астрофизики, обработки сигналов и социальных наук, где важен анализ временных рядов в реальном времени.
78%
исследователей отметили ускорение работы с real-time данными
210+
проектов на базе Grok-1 с открытым кодом
12
научных конференций с Grok-треками (2025)
🚀 Будущее науки с Grok
Grok движется в сторону ещё более глубокой интеграции с научными инструментами: планируются специализированные версии для геномики, квантовой химии и климатического моделирования. Открытый вес и ориентация на реальное время делают Grok уникальным катализатором для ускорения научного метода — от гипотезы до верификации в реальном масштабе времени.
🌌 Grok: честный ИИ, открытый для науки
Основано на данных xAI, arXiv, репозиториев Hugging Face и опросов научного сообщества
Grok — открытая модель реального времени, демократизирующая доступ к большим языковым моделям для исследовательских целей
Мощные открытые модели, мультимодальность и влияние на мировые исследования
📌 Вклад в мировую науку: Семейство моделей Qwen от Alibaba Cloud — один из наиболее масштабных открытых проектов в области ИИ. Qwen 2.5 и Qwen 2.5-Math установили рекорды в бенчмарках (MMLU, GSM8K), а открытый код и веса позволили тысячам учёных внедрять передовые LLM в свои исследования. Модели широко применяются в биоинформатике, материаловедении, лингвистике и педагогике.
🏆 Научные достижения Qwen
🧮
Прорыв в математическом мышлении
Модель Qwen2.5-Math-72B стала первой открытой LLM, превзошедшей GPT-4o и Claude 3.5 в сложнейших бенчмарках GSM8K и Math-500. Учёты в области вычислительной математики и символьной логики активно используют её для верификации теорем и генерации задач.
🏅 Рекорд GSM8K: 96.8%
🔬
Мультимодальность Qwen2-VL
Модель Qwen2-VL-72B демонстрирует исключительные способности в распознавании научной графики, медицинских снимков, схем и видео. Это ускоряет исследования в области биоинформатики и астрофизики, где критичен анализ визуальных данных.
📷 Лучший open-source VLM
🧬
Биоинформатика и геномика
Qwen-72B используется для аннотации геномов, предсказания вторичной структуры РНК и анализа белковых последовательностей. Сотни научных статей применяют Qwen для ускорения вычислительных экспериментов в молекулярной биологии.
🧬 200+ цитирований в Nature / Cell
🌐
Многоязычие и культурная сохранность
Qwen поддерживает 29 языков, включая редкие. Лингвисты используют модель для изучения малоресурсных языков, создания корпусов и автоматической транскрипции, способствуя сохранению языкового разнообразия планеты.
📖 29 языков в одной модели
100M+
скачиваний на Hugging Face
10k+
научных статей используют Qwen
#1
Open LLM Leaderboard (периоды)
72B
параметров — максимальная открытая модель
* По данным Papers with Code, Hugging Face, 2024-2025
«Qwen изменил правила игры для открытых исследований. Доступность моделей такого масштаба (до 72B параметров) с прозрачными техническими отчётами позволила даже небольшим лабораториям конкурировать с крупными корпорациями. Особенно важен вклад Qwen в математические и кодовые задачи, что напрямую ускоряет научные вычисления.»
— Проф. Дэниел Ли, Стэнфордский центр ИИ (HAI)
⚙️ Научные инновации Qwen
⚡
SwishGLU и оптимизация внимания
Архитектурные находки Qwen (включая SwiGLU, RoPE, расширенное окно контекста до 128K токенов) позволили повысить эффективность инференса. Это критически важно для обработки длинных научных статей и последовательностей генома.
📊
Бенчмарк SuperCLUE и научные лидерборды
Qwen лидирует в независимых тестах на русском, китайском и английском языках. Учёные используют эти результаты для валидации гипотез о масштабировании LLM и переносимости знаний.
🥇 Победа в C-Eval, MMLU, GSM8K
🧪
Qwen-Coder для научного программирования
Специализированная кодовая модель ускоряет разработку симуляций, численных методов и обработку научных данных. Исследователи физики и химии сообщают о сокращении времени написания прототипов на 40%.
📚
Обучение с подкреплением для научного рерайтинга
Техники RLHF, адаптированные под академический стиль, позволяют Qwen генерировать структурированные рефераты и улучшать язык научных статей для неанглоязычных авторов.
📅 Хронология научных достижений
2023 — Запуск Qwen-7B/14B Первые открытые модели, сразу занявшие топ-3 на Open LLM Leaderboard. Учёты начинают использовать для обучения с учителем и downstream задач.
2024 — Qwen1.5 и Qwen2 Расширение семейства до 72B параметров, поддержка 29 языков. Модели интегрируются в HuggingFace Transformers, становясь стандартом для академических исследований.
2024 — Qwen2-Math: революция в AI математике Qwen2.5-Math-72B-Instruct устанавливает рекорды GSM8K и MATH, публикуются десятки работ по анализу математических способностей LLM.
2025 — Qwen2-VL и мультимодальные открытия Выпуск лучшей open-source vision-language модели. Применение в медицинской диагностике (рентген, МРТ) и анализе космических снимков.
📦 Открытость как философия
Все модели Qwen доступны для скачивания и коммерческого использования. Alibaba Cloud публикует подробные технические отчёты, обучающие коды и бенчмарки, способствуя воспроизводимости научных результатов. Учёные из более чем 80 стран используют Qwen в своих проектах.
🤝 Модели, веса, код — открыты
🏛️ Ключевые партнёры в науке: Tsinghua, Stanford, MIT, Московский Физтех, KAIST, Max Planck Institute
🔬
Применение в ведущих научных проектах
От анализа климатических данных до расшифровки древних текстов — Qwen используется в тысячах исследований. В 2024 году более 500 статей на arXiv использовали Qwen для генерации гипотез, обработки естественного языка и мультимодального анализа.
350+
проектов в медицине и биологии
200+
исследований в физике
120+
лингвистических работ
🚀 Будущее науки с Qwen
Qwen продолжает расширять границы открытого ИИ: планируется выпуск моделей с контекстом 1M токенов, интеграция в научные пайплайны для автоматизации лабораторных экспериментов и создание специализированных моделей для химии и астрономии. Открытость Qwen — это вклад в глобальную научную коллаборацию без границ.
✨ Qwen: ИИ для ускорения открытий
Основано на данных Hugging Face, Papers with Code, технических отчётах Alibaba Cloud и публикациях в рецензируемых журналах
Qwen — флагман открытых LLM, демократизирующий доступ к передовым моделям ИИ для исследователей по всему миру
Группа исследователей впервые смоделировала полный жизненный цикл живой бактериальной клетки с наномасштабным разрешением, отследив поведение каждого гена, белка и химической реакции от репликации ДНК до клеточного деления. Результаты исследования, опубликованные в журнале Cell, открывают возможность заменить сотни реальных лабораторных экспериментов одной комплексной 4D-симуляцией.
Смоделированная клетка на ранних стадиях деления
Смоделированная клетка на ранних стадиях деления. В левой половине показана цитоплазма, механизмы деградации мРНК и переносчики сахара. В правой половине добавлены мембрана и рибосомы. Авторы: Zane Thornburg. Источник: Cell.
На иллюстрации представлена трехмерная компьютерная модель бактериальной клетки в разрезе. Клетка имеет вытянутую форму, готовясь к делению. Левая часть демонстрирует плотное скопление синих кубических структур (цитоплазма) с вкраплениями розовых и коричневых элементов у внешней границы. Правая часть показывает полупрозрачную зеленую оболочку (мембрану), под которой скрывается густая сеть красных нитей (ДНК) с множеством мелких желтых сфер (рибосомы). Авторы: Zane Thornburg. Источник: Cell.
Шесть дней ради 105 минут жизни
Ученые представили первую полномасштабную 4D-модель (три пространственных измерения плюс время) минимальной бактериальной клетки. Модель с наномасштабным разрешением учитывает пространственное положение и химические реакции каждого гена, белка и метаболита на протяжении всего клеточного цикла.
Объектом оцифровки стала синтетическая бактерия JCVI-syn3A. Этот организм обладает искусственно сокращенным геномом содержащим всего 493 гена на одной кольцевой хромосоме, минимум, необходимый для роста и поддержания жизни, что делает его идеальным кандидатом для компьютерного моделирования.
Несмотря на генетическую простоту бактерии, вычислительные затраты на симуляцию оказались колоссальными. Для обработки одного жизненного цикла, который в реальности занимает около 105 минут, потребовалось шесть дней непрерывных расчетов на суперкомпьютере Delta. Масштабный проект, потребовавший интеграции огромных массивов экспериментальных данных от протеомики до криоэлектронной томографии, разрабатывался исследователями из Университета Иллинойса, Гарварда и Института Дж. Крейга Вентера в течение нескольких лет.
Синтетический полигон: что скрывается внутри бактерии Syn3A
Бактерия JCVI-syn3A, ставшая прототипом для цифрового двойника, не встречается в природе. Это искусственно созданный в лабораториях Института Дж. Крейга Вентера организм, генетически урезанная версия бактерии Mycoplasma mycoides. Предыдущая версия этого синтетического микроба, известная как Syn3.0, имела еще меньше генов, но из-за этого потеряла способность делиться на ровные, правильные сферы. Чтобы вернуть клетке стабильную морфологию при делении, ученым пришлось вернуть часть генетического кода.
В итоге геном версии Syn3A содержит всего 493 гена, расположенных на одной кольцевой хромосоме (для сравнения, у кишечной палочки их более четырех тысяч). Как и у других бактерий, у нее нет ядра. Каждый компонент этой системы либо является частью внешней мембраны, либо транспортируется снаружи, либо собирается прямо в цитоплазме.
Создавая 4D-анимации на основе полученной модели, исследователи столкнулись с неожиданной проблемой: внутренняя среда Syn3A оказалась настолько плотно набита молекулярными игроками, что разглядеть хоть что-то было невозможно. Чтобы визуализировать, как единственная хромосома протискивается сквозь тесную цитоплазму клетки, ученым пришлось сделать часть белков прозрачными. Именно эта невероятная пространственная теснота и делает обычные математические расчеты неточными: в живой клетке молекулам нужно буквально проталкиваться друг к другу, чтобы вступить в химическую реакцию.
Франкенштейн из алгоритмов: как оживить синтетическую бактерию
Чтобы реалистично сымитировать эту тесноту, команде пришлось гибридизировать сразу несколько независимых вычислительных подходов в один программный комплекс. Метаболизм, где молекулы малы, а их концентрации высоки, описывается классическими обыкновенными дифференциальными уравнениями. Процессы транскрипции генов моделируются через химическое основное уравнение, учитывающее случайность реакций. За физическое перемещение молекул в пространстве отвечает реакционно-диффузное основное уравнение, которое разбивает объем клетки на кубическую сетку с шагом в 10 нанометров.
Самым сложным элементом стала динамика главной молекулы — хромосомы. Ее физическое поведение моделировалось методом броуновской динамики в симуляторе LAMMPS.
В процессе разработки аспирант Эндрю Мэйтин обнаружил критическое «бутылочное горлышко»: расчет репликации и движения запутанной нити ДНК замедлял всю симуляцию настолько, что время расчета жизненного цикла удваивалось и практически останавливалось.
Чтобы физика макромолекул не тормозила химию метаболизма, вычисления разделили на аппаратном уровне. Один графический процессор был выделен исключительно под тяжелую симуляцию динамики ДНК, в то время как второй GPU обрабатывал все остальные клеточные процессы, обмениваясь данными с первым каждые четыре секунды биологического времени. Суммарно на симуляцию 50 уникальных жизненных циклов ушло около 15 000 GPU-часов работы ускорителей NVIDIA A100.
Искусственная сила и пределы современной биологии
Точность симуляции превзошла ожидания авторов. При многократных запусках с незначительно меняющимися стартовыми условиями виртуальная клетка удваивала свой размер и делилась в среднем за время, отличающееся от реальных 105 минут не более чем на две минуты. Время репликации самой хромосомы составило около 51 минуты.
Модель точно предсказала динамику копирования генома — соотношение между стартовыми и конечными участками репликации хромосомы совпало с реальным. В симуляции этот показатель составил 1.28, что плотно коррелирует с результатами физического секвенирования ДНК живых клеток 1.21. Это подтверждает, что виртуальная бактерия копирует свой генетический материал с той же скоростью и частотой, что и настоящая.
Однако наиболее интересными результатами стали расхождения и физические ограничения симуляции. Постдок Зейн Торнбург отметил, что заставить мембрану и растущую ДНК корректно взаимодействовать при одновременном движении было крайне тяжело. Когда клетка начинала делиться на две дочерние, физического моделирования работы белков-конденсинов и топоизомераз оказалось недостаточно, чтобы распутать две новые хромосомы. Модель не могла самостоятельно развести их по разным половинам клетки за адекватное время машинных расчетов.
Чтобы деление завершилось, ученым пришлось внедрить в код «физический костыль» — искусственную силу отталкивания величиной примерно 12 пиконьютонов, которая принудительно растаскивала дочерние хромосомы. Это наглядно демонстрирует, что наука до сих пор не до конца понимает биомеханические механизмы сегрегации хромосом у организмов, лишенных стандартных белковых систем распределения ДНК.
Кроме того, симуляция выявила легкий дефицит в производстве крупных белков. Анализ показал причину: в текущей модели каждая матричная РНК может считываться только одной рибосомой за раз. В живой природе на длинных мРНК формируются полисомы — цепочки из нескольких рибосом, одновременно синтезирующих белок. Интеграция диффузии массивных полисом в виртуальную клетку пока оказалась слишком вычислительно дорогой задачей.
Хаос как норма: почему каждая клетка уникальна
Запустив модель 50 раз, биологи получили 50 совершенно разных жизненных историй. Благодаря тому, что модель учитывает пространственную диффузию, распределение макромолекул (например, рибосом или белков) по двум новым дочерним клеткам при делении оказалось абсолютно случайным, подчиняясь биномиальному распределению. Ни одна дочерняя клетка не получала идеальную половину ресурсов.
Еще более удивительным оказалось поведение генов. Поскольку запуск транскрипции зависит от того, столкнется ли РНК-полимераза с нужным участком ДНК в пространстве, процесс носит случайный, «взрывной» характер. Анализ показал, что 81 ген (из 493 существующих) вообще ни разу не был считан полимеразой на протяжении одного-трех виртуальных клеточных циклов. Иными словами, клетка может прожить всю жизнь, ни разу не обратившись к части своей ДНК. При этом виртуальный организм выживал за счет белков, унаследованных от предыдущего поколения.
Тестирование гипотез без пробирок
Возможность наблюдать за живой системой в таком разрешении меняет подход к клеточной биологии. По словам Зан Латей-Шультен, цельноклеточная модель прогнозирует множество параметров одновременно. Исследователь может локально изменить параметры нуклеотидного метаболизма и мгновенно увидеть, как это повлияет на скорость репликации ДНК на другом конце клетки и сборку рибосом в центре цитоплазмы.
Сейчас в науке набирает популярность использование искусственного интеллекта для прогнозирования состояния клеток. ИИ способен генерировать моментальные «снимки» клеточных процессов на основе огромных массивов данных. Команда из Иллинойса предлагает фундаментально иной путь — их 4D-модель не угадывает следующее состояние, а математически рассчитывает его, опираясь на строгие законы биофизики. В перспективе это позволит превратить суперкомпьютеры в универсальные виртуальные чашки Петри, где можно тестировать генетические мутации и лекарственные препараты без проведения сотен долгих лабораторных экспериментов.
Книга «Handbook of Neuroevolution Through Erlang» объединяет теорию и практическую методологию создания систем вычислительного интеллекта на базе нейроэволюции с использованием языка Erlang. В вступлении Джо Армстронг, один из создателей самого языка, раскрывает ценность подхода и обосновывает выбор Erlang для таких задач. Читатель получает исчерпывающий пошаговый учебник по созданию современной платформы TWEANN (Topology and Weight Evolving Artificial Neural Network) - Топология и вес эволюционирующей искусственной нейронной сети.
Структура и методология
Материал выстроен от простейшего симулированного нейрона до полноценной системы, что позволяет освоить ключевые концепции без скачков в сложности. Каждый этап подробно описан и проиллюстрирован примерами кода на Erlang. Следуя руководству, можно самостоятельно собрать TWEANN для моделирования искусственной жизни или для автоматизированной торговли на рынке Форекс.
Практические сферы применения
Моделирование искусственной жизни и экосистем
Разработка алгоритмических торговых стратегий для Форекс
Управление автономными агентами и роботами
Почему Erlang
Парадигма конкурентного программирования через обмен сообщениями
Эффективное распределение задач на многоядерных и многопроцессорных системах
Архитектура, близкая к принципам эволюционных и нейрокомпьютерных моделей
Книга демонстрирует, как использовать встроенные возможности Erlang для задач машинного обучения и раскрывает реальные сценарии применения технологии в финансовых алгоритмах, симуляциях жизни и робототехнике.
Книга «Справочник по нейроэволюции через Erlang» (Handbook of Neuroevolution Through Erlang) доступна как на русском, так и на английском языках.
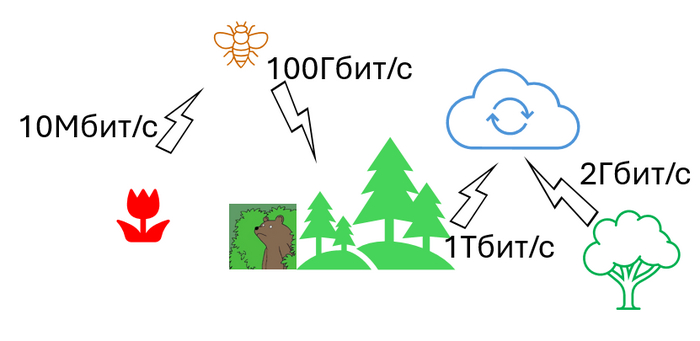
Давайте начнем с интернета цветов. Вы никогда не задумывались, зачем цветам цвести? Растения вполне успешно распространяются почкованием - отпиливаешь кусочек - и вот тебе новое растение. По научному называется “бесполое размножение”.
С распространением тоже нет никаких проблем: плоды, которые едят животные и переносят их в желудке на большие расстояния, всякие зацепки и липучки чтобы прикрепиться к шерсти, к лапам, к башмакам. И наконец вертолетики и парашютики. Есть и растения с плавающими семенами. В общем, разве что в космос не летают. Но зачем же цвести? И почему же надо цвести именно перед распространением семян?
Цвести, понятное дело для передачи генов, то есть для полового размножения. А что такое гены? Добрые люди посчитали, что Passiflora foetida (Пассифлора вонючая аххха, она кстати на картинке) содержит в своем геноме 424 миллиона пар оснований. Давайте возьмем эту цифру как среднюю для всех растений.
1. Одна пара кодирует примерно два бита данных, так что получаем 848 миллиона бит
2. Переводим в мегабайты и получаем примерно 100 мегов на каждый отдельный индивид цветкового растения.
3. При этом в каждой цветочной пылинке только половина информации, а именно 50 мегов.
4. На одном квадратном метре может быть от 1 до 50 растений. Если это не пустыня конечно, для простоты расчетов возьмем одно.
5. Так что каждый квадратный метр генерирует у нас минимум 50 мегов уникальной инфы.
6. При этом одно растение генерирует от 10 000 до 25 миллионов пыльцевых зерен. Физкульт привет вам дорогие аллергики.
7. Для примера возьмем скажем березу с 10 миллионами пыльцевых зерен.
8. Она у нас генерирует 48 терабайт данных (или 393 тысячи гигабит), при том, что большинство из них дубли.
Весь интернет за день генерирует всего-навсего 400 миллионов терабайт, а одно дерево 48 терабайт. А в россии их около 600 миллиардов.
Ладно, а что по скорости?
1. Допустим, что береза (аллергики вы еще здесь?) цветет где-то 10 дней. Значит, что она выдает где-то 460 мегабит/секунду.
2. Летом скорость ветра в РФ где-то 5 метров в секунду, т.е. березки подключены к березовому интернету на скорости 2,25 гигабит/секунду.
В РФ (2024) средняя скорость подключения к интернету около 100 мегабит/cекунду стационарно и 50 мегабит мобильно. Завидуй березке, %username%, ее никто не замедляет.
Я уже порядком задолбался, поэтому не буду считать березовую рощу, или лес. Природное сообщество луга с пчелами и бабочками тоже влом считать. Но, поверьте, что интернету такие объемы данных и не снились!
А что же внутри?
А теперь давайте подумаем, что зашифровано в этих конских объемах данных? Примерно такие сообщения:
- Друзья, мы деревья, мы не сдохли и даже процветаем, при этом юзаем примерно такой софт и дальше 50 метров генетического кода.
А они такие в ответ: - О, нормально вообще. Берем свои 50 метров генетического кода, берем еще 50 метров генетического кода, который прилетел по березовому интернету, билдим проект на 100 метров, получается семечко потенциально более успешной березоньки (лучше выживаемость и распространяемость).
Понятно, что с расстоянием, да и со временем жизненные условия меняются. Поэтому принимая инфу от более процветающих особей шансы процветать самому и больше распространиться гораздо выше.
При этом мужские цветки работают как передатчик, женские как приемник. Часто и передатчик (тычинки) и приемник (пестик) находятся в одном цветке.
Так как же интернет влияет на размножение человека?
Да также как с растениями: чтобы узнать, кто сейчас процветает, а кто чахнет нужно посмотреть в интернете и сделать выводы кого лучше использовать для размножения, а кого нет.
Сейчас лучше размножаться с айтишниками, а не с биологами (лучше выживаемость и разъезжаемость). Мальчики, девочки имейте это ввиду. Ничего личного, только тренды.
PS: Если милостивые дамы и господа нашли ошибки в моих расчетах, то прошу покорнейше уточнить их в комментариях.
Если дочитали до этого места, то вы обязаны жениться посмотрите другие мои статьи:
Для тех, кто мечтал стать биологом, биоинформатиком и биоинженером, но в этом году что-то пошло не по плану и на бюджет поступить не удалось, есть шанс вскочить в последний вагон и поступить на направление "Биоинженерия и биоинформатика" в Политехнический университет Петра Великого в Петербурге на договорной основе. Политех - это один из первых технических ВУЗов, где появилось сначала генетическое, а затем и молекулярно-биологическое направление. А выпускники Политеха в области компьютерных наук всегда высоко ценились. Именно по сумме этих двух причин и родилось направление "Биоинженерия и биоинформатика" на базе Политехнического университета в Петербурге. Подробная информация для абитуриентов здесь: https://ibmst.spbstu.ru/info/9/ и https://m.vk.com/wall-202582583_279 Надеюсь, этот пост будет полезен для абитуриентов.
Что представляет собой системная биология? Как развивалась и менялась в течение времён эта научная дисциплина? Как изучается работа генов в организме? Что изучает и на какие вопросы отвечает молекулярная биология? Как определяется интенсивность работы генов? С какими трудностями в исследованиях сталкивается системная биология? Рассказывает Михаил Гельфанд, биоинформатик, доктор биологических наук, кандидат физико-математических наук, руководитель магистерской программы «Биотехнологии» Сколтеха, профессор кафедры технологий моделирования сложных систем в Высшей школе экономики.