

Влияние массажа на мОЛЕКУЛЯРНОМ уровне
Для размышления и не более
Для размышления и не более

Израильские ученые из Университета Бар-Илан представили результаты уникального исследования, в ходе которого им удалось изменить пол мыши, внеся точечную правку в так называемую "темную материю" генома. Как сообщает издание The Times of Israel, это первый в мире случай, когда замена всего одной "буквы" в некодирующем участке ДНК привела к радикальной смене биологического развития организма.
Традиционно считалось, что за развитие пола отвечают исключительно гены, кодирующие белки. Однако команда под руководством доктора Ницан Гонен из факультета наук о жизни и Института нанотехнологий сфокусировалась на 98% генома, которые раньше называли "мусорной ДНК", поскольку они не участвуют в синтезе белков. Используя технологию генетических ножниц CRISPR, исследователи изменили работу переключателя Enh13, который контролирует ген Sox9.
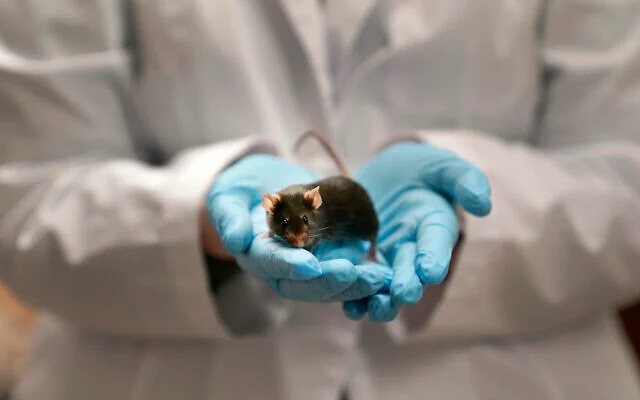
"Мы заменили всего одну букву ДНК из примерно 2,8 миллиарда в месте, которое ничего не кодирует. Я была совершенно удивлена, обнаружив, что мыши с генотипом XX, которые генетически должны были быть самками, развились в самцов с полноценными яичками и мужскими половыми органами", - пояснила доктор Ницан Гонен. По ее словам, изменение в этом специфическом участке позволило предотвратить формирование яичников, запустив вместо них развитие мужской репродуктивной системы.

Доктор Ницан Гонен из факультета наук о жизни им. Мины и Эверарда Гудмана и Института нанотехнологий и передовых материалов Университета Бар-Илан.
Результаты рецензируемого исследования, проведенного под руководством аспирантки и ведущего автора Элишевой Аббербок и других исследователей из Университета Бар-Илан, а также доктора Ариэля Афека из Института Вейцмана и доктора Фрэнсиса Пула из Университета Монпелье, были опубликованы в четверг в журнале Nature Communications.
Это открытие имеет колоссальное значение для современной медицины и биологии. Оно доказывает, что "темная материя" ДНК, составляющая подавляющую часть нашего генетического кода, играет определяющую роль в развитии организма и возникновении различных заболеваний. Ученые полагают, что понимание механизмов работы этих некодирующих участков поможет объяснить природу многих генетических мутаций, причины которых ранее оставались загадкой для науки.
Перевод с английского

Наша история о том, как снежных барсов помогают сохранять... Экскременты!
В современной сфере охраны природы для изучения животных, помимо традиционных методов — тропления по следам, визуального наблюдения или автоматических камер, давно используют и молекулярно-генетический анализ.
Для этого учёные собирают образцы шерсти и экскрементов во время экспедиций, зоопарках и питомниках.


Специалисты Стенфордского университета изучили 41 образец ДНК — 35 ирбисов из дикой природы и 6 из зоопарков.

Исследование доказало: снежные барсы генетически близки друг к другу намного больше, чем другие виды больших кошек.
Причина такого низкого генетического разнообразия — тот факт, что популяция ирбисов долгое время была стабильной, а её численность — низкой.
Скорее всего, особи с вредными мутациями просто погибали до того, как могли оставить потомство, в может нежизнеспособными были уже их отпрыски. Популяция проходила определённую «очистку» и сохраняла относительное здоровье.
Такое низкое генетическое разнообразие популяции, к сожалению, увеличивает уязвимость вида перед угрозами.
Выводы учёных лишний раз подтверждают: ирбисы нуждаются в эффективной охране с учётом растущего антропогенного прессинга на хрупкую экосистему гор.

Южная Африка намерена стать первой страной, включившей раздел о наследственном или зародышевом редактировании генома человека в свои руководящие принципы этики медицинских исследований, что вызвало бурную реакцию научного сообщества.
В обновленном документе подробно изложены условия для проведения исследований в области геномного редактирования. В частности, подчеркивается важность строгого этического контроля, научного обоснования и мониторинга здоровья людей, родившихся с генетическими изменениями. Ученые обеспокоены тем, что в ЮАР могут разрешить модификацию генов в яйцеклетках, сперматозоидах или эмбрионах, что является практикой, которая до сих пор запрещена в других странах.
В документе указано, что такие исследования должны иметь четкое научное обоснование и быть направлены исключительно на предотвращение серьезных заболеваний. Процедура требует прозрачности, информированного согласия всех участников и строгого этического контроля. Любое вмешательство в геном человека должно быть тщательно взвешено, а потенциальные выгоды для общества должны перевешивать все риски.
Исследователи обязаны проводить долгосрочный мониторинг здоровья и благополучия людей, рожденных в результате наследственного редактирования генома. Цель этого мониторинга — выявление любых непредвиденных последствий генетических изменений. Генетики также должны соблюдать все соответствующие законы, регулирующие такие исследования.
Ни одна страна не разрешает явное редактирование генома человека в клинических условиях. Международное научное сообщество единогласно выступает против этой практики. Хотя технология открывает перспективы для лечения наследственных заболеваний, таких как муковисцидоз и серповидноклеточная анемия, она также поднимает множество этических вопросов и создает опасность злоупотребления.
Некоторые опасаются, что она может привести к одному из проявлений технофашизма, что вызывает беспокойство у технофобов и тех, кто испытывает футурошок — иррациональный страх перед новыми возможностями и изменениями в привычном укладе жизни.
В 2018 году китайский биофизик Хэ Цзянькуй, работавший в Южном университете науки и технологий в Шэньчжэне, сделал сенсационное заявление: он утверждал, что создал первых в мире генетически модифицированных детей. Его слова вызвали бурную реакцию во всем мире.
В 2020 году Хэ Цзянькуй был приговорен к тюремному заключению за нарушение медицинского законодательства. Однако в апреле 2022 года его освободили, и он смог вернуться к научной деятельности.
В 2019 году международное сообщество ученых и этиков призвало ввести мораторий на клиническое применение технологии редактирования генома человека, передающегося по наследству. Это предложение получило поддержку Национальных институтов здравоохранения США. В 2023 году участники третьего международного саммита по редактированию генома человека подтвердили, что такая практика остается неприемлемой.
Существуют разные точки зрения относительно того, действительно ли южноафриканские законы допускают наследственное редактирование генома человека. Некоторые учёные считают, что, несмотря на обновленные руководящие принципы исследовательской этики, законодательство ЮАР по-прежнему запрещает клиническое применение наследственного редактирования генома человека.
Другие утверждают, что существующее законодательство уже допускает такую процедуру, а обновленные руководящие принципы лишь отражают эту реальность и предоставляют рекомендации. Исследователи также обеспокоены тем, что новый документ может быть использован для дальнейшей легализации создания генетически модифицированных детей, и призывают к более детальному изучению причин такой формулировки рекомендаций.
#eofru #высокие_технологии #биотехнологии #генетика #трансгуманизм #медицина_будущего #биопанк
В этой лекции по криминалистике. Что из себя представляют следы рук? Какие перспективы имеются у ДНК-идентификации следов рук? Об особенностях обнаружения и сбора следов рук, особенностях ДНК-исследования следов рук и современных подходах к ДНК-анализу следов рук рассказывает Татьяна Фалеева, врач – судебно-медицинский эксперт отделения молекулярно-генетической идентификации филиала №2 ФГКУ «111 Главный государственный центр судебно-медицинских и криминалистических экспертиз» Министерства обороны РФ.
В этой лекции по криминалистике: что является объектами ДНК-экспертизы? Какие существуют преимущества ДНК-методов перед стандартными методами судебной биологии? Возможны ли ошибки при проведении ДНК-экспертизы? Каков современный опыт ДНК-идентификации древних останков и перспективы развития методов судебной генетики? О проблемах и перспективах ДНК-идентификация личности рассказывает Игорь Валериевич Корниенко, доктор биологических наук, главный научный сотрудник Южного научного центра РАН, заведующий научной лабораторией «Идентификация объектов биологического происхождения» Академии биологии и биотехнологии им. Д. И. Ивановского ЮФУ.
Какими были первые случаи использования экспертизы ДНК для раскрытия преступлений в криминалистике и для определения родства? Изобретение техники ДНК-дактилоскопии британским генетиком Алеком Джеффрисом. Изобретение метода Полимеразной цепной реакции (ПЦР) американским биохимиком Кэри Муллисом и новые возможности, которые он дал криминалистике, археологии и другим областям знаний. Что такое секвенаторы ДНК? Базы данных ДНК для судебно-медицинской идентификации в уголовных делах. Рассказывает Игорь Корниенко, генетик, доктор биологических наук, заведующий лаборатории идентификации объектов биологического происхождения Академии биологии и биотехнологии ЮФУ, главный научный сотрудник Южного научного центра РАН.
ВКонтакте: https://vk.com/video-190320587_456240067
Чтобы ответить на этот вопрос, в научном исследовании необходимо определиться с целью, задачами и методами, и изучаемыми материалами. Для этого нужно постараться предварительно поставить гипотезу, которая облегчит нам понимание того, чего мы хотим, а следовательно, позволит нам выбирать материалы исследования.

В качестве гипотезы можно опираться на ваши знания в области классификации групп животных. Однако, если у Вас нет таких знаний и Вы не хотите страдать в поисках этих знаний в полях, лесах и лабораториях, то Вы можете стать продвинутым пользователем интернета и воспользоваться удобном сайтом lifemap [1], который отображает филогенетическое древо всех животных. Если же вы не продвинутый пользователь, то Вы можете просто воспользоваться википедией. Стоит отметить, что для учёного сайт Lifemap является таким же примитивным, как и википедия, но не бойтесь начать с малого, ведь википедия может послужить толчком к эволюции от простого к сложному. Поэтому пойдёмте эволюционировать на вики вместе. Для этого зайдём в поисковик и посмотрим информацию о нужных нам группах, с которыми в будущем нам предстоит работать, на данном сайте. Первые в списке у нас медвежьи. На странице сайта нам не нужно досконально изучать строение, размножение и образ жизни медведей. Нам нужны три вещи:

Переходим в раздел научной классификации и смотрим список родов в семействе медведей, предварительно выписав название этого семейства на латыни (Ursidae). Нам понадобятся названия всех родов на латыни, которые есть в семействе. Их лучше также выписать (рис.1).
(рис.1)

После проделанной работы переходим в раздел филогенетики и выбираем кладу с ближайшими живыми родственниками в качестве запасного варианта.
Это нужно сделать на случай, если нужные генетические последовательности медведей из разных родов в генбанке нам найти не удастся (рис.2).

Нам повезло, по альтернативной версии ближайшие родственники — это ластоногие. Выпишем название этой группы, выберем семейство и список родов аналогично медведям.
Теперь переходим в раздел краткой сводки научной классификации (рис.3). Находим вкладку «отряд хищные» и переходим по ней.
(рис.3)
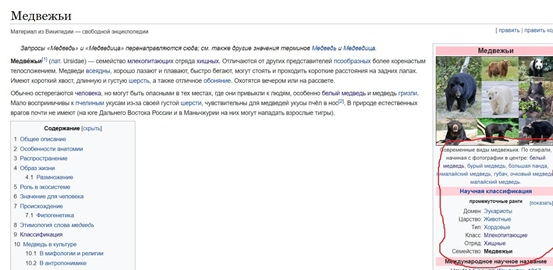
Во вкладке отряда переходим в раздел 4.1. «Внешняя систематика». Там необходимо найти надотряд, к которому принадлежат медведи (рис.4). Он указан на филогенетическом древе в виде гиперссылки, нажимаем на неё и переходим в соответствующий раздел.
(рис.4)
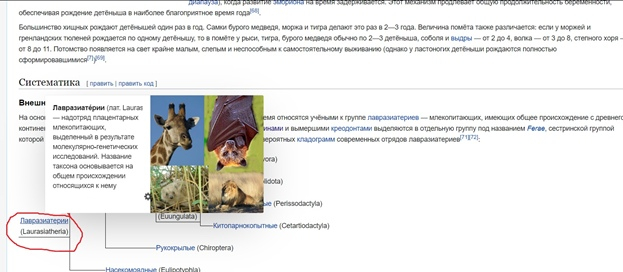
В разделе нам нужно узнать отношение приматов к этому надотряду. Для этого мы переходим во вкладку «классификация» и как ни странно мы не обнаруживаем в нём приматов (рис.5) Получается, что медведи по версии википедии вообще не близкие родственники приматов. Может это так и есть, но кто же по версии википедии примату будет братом?
(рис.5)

Для этого проводим аналогичные манипуляции с семейством хомяковые и в конечном итоге попадаем в отряд грызуны. Переходим в раздел систематики и ищем надотряд (рис.6).
(рис.6)
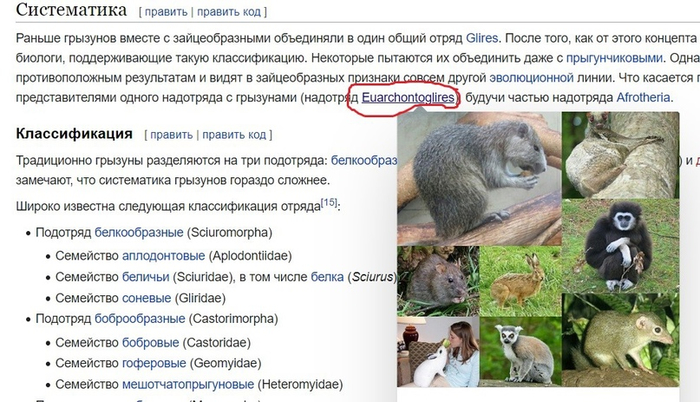
Какое удивление! Мы в надотряде Euarchontoglires обнаружили людей! Чудесно. Ну, а теперь можно поставить гипотезу. Нашей гипотезой будет утверждение, что хомяковые являются братьями людей, а нашей целью подтвердить это утверждение. Для достижения цели нам необходимо поставить следующие задачи:

Чтобы уже начинать определяться с методами нам нужно выбрать внешнюю группу, а также определиться с генетическими последовательностями. Внешняя группа нам нужна для определения положения корня дерева, так сказать, его основы. В качестве внешней группы обычно используют одну или несколько клад, отпочковавшихся от общего дерева заведомо раньше (но желательно ненамного раньше) анализируемых последовательностей. Поскольку мы изучаем филогенетические отношения между плацентарными млекопитающими, то в качестве внешней группы можно использовать сумчатых млекопитающих [4]. Пусть это будут опоссумы. Я люблю опоссумов. Это котики мира сумчатых, а котиков любят все.
Далее определяем материалы. В качестве необходимых материалов я решил взять рибосомальные гены 18S рРНК у двух представителей разных родов из каждых изучаемых групп. Маркер 18S рРНК используется с конца 70-х годов прошлого столетия и является универсальным для систематических построений. Ген, кодирующий 18S рибосомную РНК, есть в геноме всех известных эукариот и является удобным маркером для их идентификации; он отсутствует у вирусов, бактерий и архей. Ген 18S рРНК содержит как консервативные участки, одинаковые у всех прокариот, так и вариабельные. Консервативные участки служат для первого этапа полимеразной цепной реакции – присоединения праймеров к исследуемой ДНК-матрице, вариабельные участки – для идентификации видов. Степень сходства видоспецифичных вариабельных участков отражает эволюционное родство разных видов [3].
С материалами более-менее определились, теперь их необходимо скачать в генетической базе данных. Переходим на сайт ген банка и в поисковой строке вбиваем название семейства латинскими буквами и ищем генетические последовательности родов, которые мы записывали ранее. Последовательности должны быть приблизительно равной длины и ни в коем случае не короткие, ибо короткие последовательности несут мало информации, а информация в нашей работе — это золото, где филогенетическое древо — Зиккурат. А всем мы знаем, что для строительства Зиккурата нужно больше золота. Поэтому для удобства в графе «Sequence length» выставим необходимую длину последовательностей (1600-2500) и нажмём кнопку «Search» (рис.7).

На рисунке выше мы видим, что я начал с медведей, к сожалению ген банк выдал мне всего три результата и все одного вида. Ничего страшного, ведь медведя мы всё равно скачаем, а запасной вариант в виде ластоногих (рис.8) у нас имеется и к счастью в генбанке необходимые последовательности по ним есть.

Таким образом мы скачиваем все необходимые нам последовательности в формате "fasta". Cкаченные последовательности закидываем по одной (или несколько, если Вы скачали всё одним форматом) в программу MEGA 10 для объединения в один формат «fasta» в будущем (рис.9)
Итак, в мою выборку исследования попали 8 видов. Я не буду пугать Вас латынью как делаю это обычно, а перечислю всех избранных товарищей по-русски «матом». Первые два вида в моём списке будут представлять этакую не существующую в реальном мире вершину эволюции и как Вы догадались это человекообразные обезьяны — человек и горилла. Вторыми по иерархии идут хомяковые — водяная полёвка и серый хомячок, третьими замыкающими внутреннюю группу идут медвежьи и настоящие тюлени — бурый медведь и длинномордый тюлень соответственно. Представляют внешнюю группу у меня два вида из разных родов опоссумов — виргинский и домовой опоссумы. Строить дерево мы будем в тренировочной программе MEGA 10
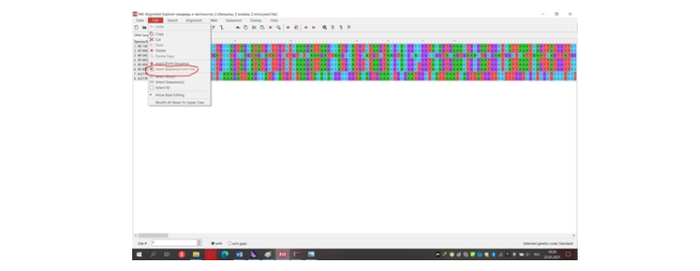
Теперь нам надо начинать определяться с методами. Первым важным методом будет выравнивание генетических последовательностей. Выравнивание является важным биоинформатическим методом, основанным на размещении двух или более генетических последовательностей позволяющим увидеть сходные участки в этих последовательностях. Их сходство может отображать структурные и эволюционные связи, которые без выравнивания не построить [5]. Выравнивание мы не будем производить в MEGA 10, так как для рибосомальных последовательностей лучше воспользоваться маффтом [6]. Перед этим мы объединим все последовательности в меге в одну и экспортируем в любую папку на рабочем столе в формате «fasta» (рис.10).

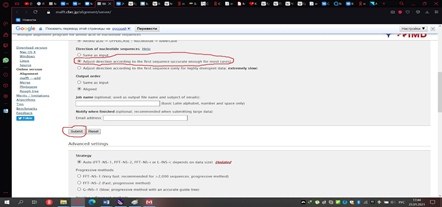
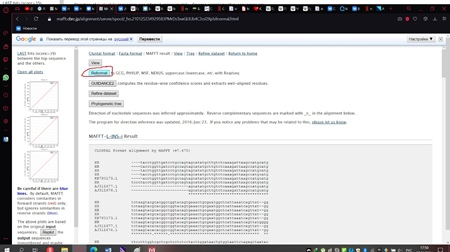
Сохранённый файл мы загружаем на сервер мафта в браузере (рис. 11) и изменим один стандартный параметр, выбрав тот, который показан на рисунке 12. Далее нажимаем кнопку «Submit» и получаем результат, который необходимо реформировать в формат fasta, как показано на рисунке 13.

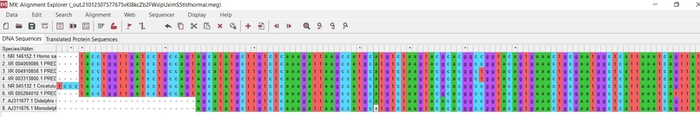
Полученный формат необходимо загрузить обратно в мегу и уже работать в ней. Поздравляю мы это сделали! (рис.14)
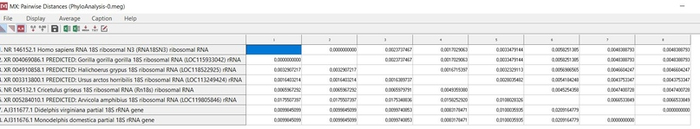
Далее производим установление попарных эволюционных дистанций между анализируемыми последовательностями, представляемых в виде матрицы дистанций. Другими словами, для построения дерева нам требуется эволюционная модель, оптимального метода расчета эволюционных дистанций между последовательностями. В качестве статистического метода я воспользуюсь методом оценки дат дивергенции видов, который разрабатывался с точки зрения концепции молекулярных часов, а именно эволюционной моделью Hasegawa, Kishino и Yano 1985 года.
Данная модель различает скорость различных точечных мутаций и учитывает не равные базовые частоты, которые не учитываются простыми моделями [7]. В меге эту модель можно выбрать сразу при построении дерева в методе максимального правдоподобия, там же заранее выставим проверку в 1000 реплик (так называемый бустрэп анализ). Данный анализ позволяет посмотреть статистическую поддержку ветвей, чем она выше, тем будет лучше. Высокая поддержка большинства ветвей более 70% позволяет сказать, что дерево построено правильно (рис.15). Поддержка ниже 70% для одной, или двух ветвей не является очень критичной при низкой выборке, но, если мы получим статистическую поддержку всех ветвей ниже 70% это будет говорить об очень плохом результате.
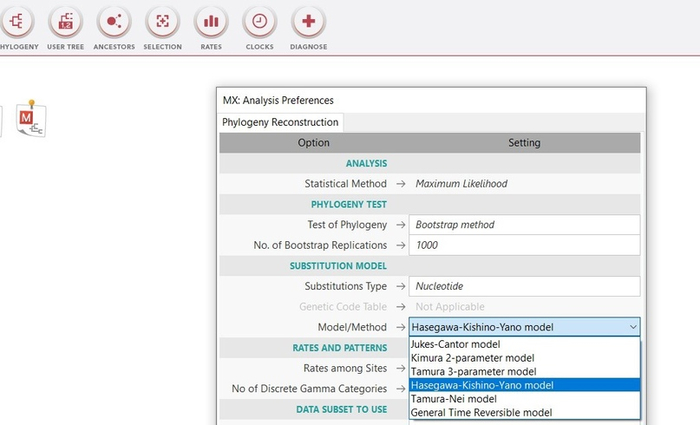
К сожалению, посмотреть эту модель отдельно инструментал меги не позволяет, но наглядно она бы выглядела примерно таким образом (рис.16).
Данная модель разрабатывалась для построения деревьев методом максимального правдоподобия, который я по сути и выбрал.
Метод максимального правдоподобия, говоря примитивным языком, позволяет определить неизвестное число параметров на основании известных результатов эксперимента. Скажем, если известно число граней правильного многогранника (т.е. число параметров), то можно определить, чему равны вероятности различных исходов бросков этого многогранника. Так, для шестигранной игральной кости вероятность любого исхода броска будет равна 1/6. Однако если взять за гипотезу, что число граней некой игральной кости нам неизвестно, данный метод позволяет предположить путём многократных повторных экспериментов в виде бросков этой игральной кости, число граней этой кости и определить правдоподобие этого предположения. Так, многократно подбрасывая некую игральную кость с неизвестным числом граней и наблюдая, что число различных исходов бросков кости равно шести, можно сделать предположение, что это кость шестигранная [4]. Именно поэтому этот метод в данном случае я считаю одним из лучших для ответа на заданные мной вопросы.
В качестве дополнительных плюшек мега позволяет воспользоваться функциями уточнения выводимого дерева, что даёт нам возможность вывести исходное дерево для эвристического поиска, который в свою очередь используется для оценки лучшего состояния нашего дерева. Подробно, что такое эвристический поиск можно прочитать в IT сообществе хабр [5]. Итак, в дополнительных параметрах меги меге мы можем выбрать метод максимальной экономии, который является критерием оптимальности, для которого наилучшим считается самое короткое дерево, которое объясняет данные. Этот метод работает по канонам Бритвы Оккама (рис 17). В принципе в дополнительных параметрах можно выбрать ещё кучу всего, но я думаю и этого вполне хватит.
Собственно, теперь у нас всё готово, чтобы проверить википедию на подлинность и заодно нашу гипотезу. Строим дерево! (Рис.18) ;(Рис.19)


Вуаля — чувствую себя доктором ВУ, когда дерево строится успешно!
Теперь давайте взглянем, что у нас получилось, а получилось у нас практически всё идеально!
Как Вы сами видите построенное дерево рассказывает нам о том, что грызуны являются более близкими родственниками по отношению к людям и подтверждает нашу гипотезу, несмотря на то, что одна ветвь у нас имеет поддержку ниже 70%, что в принципе не является критичным, так как все остальные ветви имеют статистическую поддержку более 70%. Конечно я допускаю за собой маленькие ошибки в построении дерева, но общая картина была вполне ожидаема и показала всё то, что известно самому капитану очевидности и его капитанше. Действительно хомяк является «братом» человека, а медведь его дальним родственником, а теперь можно выдохнуть! Всего доброго!
Автор: Аномалокарис, биолог, вдохновитель сообщества Фанерозой, Ефимов Самир

1. http://lifemap.univ-lyon1.fr
2. https://www.ncbi.nlm.nih.gov/nuccore/?term=Phocidae%2018S%20...
3. Соловьева В.В. Молекулярно-генетический анализ беспозвоночных животных по нуклеотидной последовательности гена 18S рибосомной РНК: учебное пособие / Соловьева В.В., Моров А.Р., Ризванов А.А., Сабиров Р.М.- Казань: федеральный ун-т, 2011 – 52 с.
4. Молекулярная эволюция и филогенетический анализ/ В.В. Лукашов —М.БИНОМ. Лаборатория знаний, 2009. — с.256. с.92-123.
5. Mount DM. Bioinformatics: Sequence and Genome Analysis. — 2nd. — Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY., 2004.
6. https://mafft.cbrc.jp/alignment/server/
Hasegawa M., Kishino H., and Yano T. (1985). Dating the human-ape split by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22:160-174.






